Actualités
Une étude sur la maladie de Huntington soutenant le concept de neuroarchéologie
La maladie de Huntington, une maladie héréditaire dominante qui se manifeste au milieu de l’âge adulte, est causée par l’expansion d’un tractus polyglutamine dans la protéine huntingtine (HTT). La mutation conduisant au tractus polyglutamine confère un gain toxique de phénotype de fonction se traduisant par une neurodégénérescence la plus sévère dans le striatum. Malgré sa manifestation symptomatique «tardive», il existe plusieurs rapports cliniques de déviations pré-symptomatiques chez des patients atteints de la maladie de Huntington montrant des altérations de la connectivité cérébelleuse-striatale1 et de la fonctionnalité du cortex préfrontal2. Dans leur publication Science3, les équipes d’Alexandra Durr et Sandrine Humbert montrent que des altérations pré-symptomatiques du neurodéveloppement dues à des mutations de la protéine HTT sont déjà présentes à 13 semaines de gestation avec des anomalies du cortex en développement, des défauts de polarité et de différenciation cellulaire, et la progression cellulaire. Les implications de ces travaux sont importantes car elles remettent en question la dissociation entre les troubles développementaux et neurodégénératifs et soulignent la nécessité d’évaluer les signatures pré-symptomatiques précoces à la recherche de traitements. Fait intéressant, cette étude s’inscrit dans le concept de Neuroarchéologie4 proposé par Yehezkel Ben-Ari il y a plus de dix ans qui suggère que les troubles neurologiques sont «nés» in utero conduisant à une persistance de neurones dotés de caractéristiques immatures qui sont la cause finale des séquelles délétères .
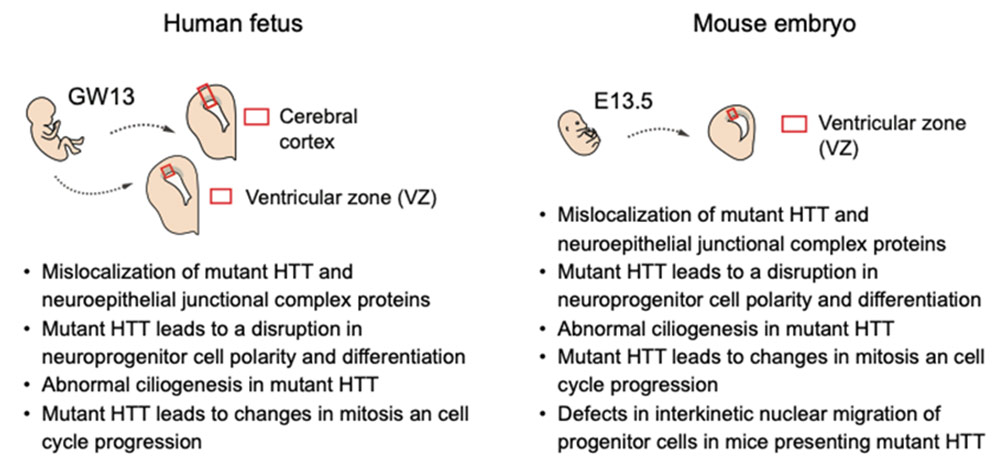
Références : 1. Tereshchenko AV. et al. Abnormal development of cerebellar-striatal circuitry in Huntington disease. Neurology. 2020, 94(18):e1908-e1915. Doi: 10.1212 / WNL.0000000000009364. 2. Wolf RC. et al. Dorsolateral prefrontal cortex dysfunction in presymptomatic Huntington’s disease: evidence from event-related fMRI. Brain. 2007, 130(11):2845-57. Doi: 10.1093 / cerveau / awm210. 3. Barnat M. et al. Huntington’s disease alters human neurodevelopment. Science. 2020, 369:787-793. Doi: 10.1126/science.aax3338. 4. Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends in Neuroscience. 2008, 12:626-36. Doi: 10.1016/j.tins.2008.09.002

Veille scientifique
- Maladie d’alzheimer: un espoir? 20 janvier 2022
- Des somnifères qui provoquent des « réveils » ? 17 février 2021
- Un nouveau rapport sur l’utilisation prometteuse du traitement par bumétanie pour l’autisme 24 janvier 2021
- Une étude sur la maladie de Huntington soutenant le concept de neuroarchéologie 16 août 2020