Essais cliniques
Qu’est-ce qu’un essai clinique ?
Un essai clinique a pour but de tester l’efficacité et les effets secondaires d’un nouveau traitement qui aura l’autorisation d’être mis sur le marché. Les essais cliniques sont précédés par des phases de recherche préclinique sur des modèles animaux et des approches in vitro afin de valider son innocuité, la liste de ses effets secondaires et son efficacité dans la réduction du syndrome. Cette procédure prend souvent de nombreuses années.
Quelles sont les différentes phases des essais cliniques ?
La recherche préclinique :
Avant d’être testé sur l’homme, un médicament potentiel ou une molécule passe par des études en laboratoire, d’abord in vitro, puis in vivo sur des modèles animaux, généralement des modèles murins. L’objectif est d’étudier ses effets – efficacité, mécanisme d’action et toxicité – à différentes échelles : au niveau cellulaire, des organes cibles, puis à l’échelle de l’organisme, incluant les aspects biologiques et comportementaux. Si les résultats sont concluants, la molécule peut entrer en phase clinique.
Phase I :
La phase I vise à évaluer la sécurité d’un médicament en étudiant son absence d’effets indésirables majeurs, et sa tolérance et son profil pharmacocinétique (absorption, distribution, métabolisme, élimination). Elle est réalisée sur un petit groupe de volontaires sains (20 à 100 participants) dans un cadre monocentrique, généralement en simple aveugle ou en ouvert, et s’étend sur quelques mois. Cette phase permet d’identifier les premiers effets secondaires et déterminer la dose optimale pour les études suivantes. L’absence d’effets secondaires majeurs conditionne la poursuite de l’essai.
Phase II :
La phase II, ou « étude pilote », a pour objectif d’évaluer l’efficacité préliminaire du médicament et d’affiner la posologie, tout en poursuivant la surveillance des effets indésirables. Elle est menée sur un groupe de 100 à 300 patients atteints de la pathologie ciblée, répartis aléatoirement en deux groupes homogènes et comparables (âge, sexe, caractéristiques de la maladie…). Un groupe reçoit le médicament à l’étude, tandis que l’autre reçoit un traitement de référence ou un placebo. L’essai se déroule en double aveugle, c’est-à-dire que ni les patients ni le personnel médical ne connaissent la nature du traitement administré. Cette phase, d’une durée de plusieurs mois à deux ans, permet de confirmer l’existence d’un bénéfice thérapeutique et de déterminer la dose optimale pour la phase III.
Phase III :
La phase III, ou « étude pivot », est l’étude comparative d’efficacité proprement dite. Elle compare le médicament à un placebo ou à un traitement existant sur un grand nombre de patients, allant de 500 à plusieurs milliers, afin de confirmer son efficacité et sa sécurité. Menée en double aveugle randomisée, cette étude se déroule dans plusieurs centres de référence (étude multicentrique) sur une période de 1 à 5 ans. L’objectif est de démontrer un rapport bénéfice/risque positif, ce qui est essentiel pour obtenir l’autorisation de mise sur le marché par les autorités compétentes.
Phase IV :
La phase IV, ou « post-marketing », correspond au suivi à long terme d’un médicament après son autorisation de mise sur le marché. Elle vise à surveiller ses effets à long terme, à détecter d’éventuels effets secondaires rares ou complications tardives, et à évaluer son efficacité en conditions réelles d’utilisation. Cette phase repose sur des études observationnelles menées sur plusieurs milliers de patients et fait partie du dispositif de pharmacovigilance, garantissant un suivi continu de la sécurité du traitement.
Neurochlore et ses essais cliniques
Comprendre les Troubles du Spectre Autistique et l’Impact des Recherches Fondamentales
Les troubles du spectre autistique (TSA) regroupent un éventail complexe de troubles affectant divers aspects du comportement, tels que les interactions sociales, la communication, la capacité d’adaptation aux changements, les comportements stéréotypés, ainsi que les intérêts restreints. Ces manifestations, pouvant être sévères et envahissantes, exercent un impact considérable au quotidien sur l’autonomie et la qualité de vie des personnes concernées, ainsi que de leur entourage. À ce jour, aucun traitement curatif n’existe pour les TSA.
Les TSA, comme de nombreuses autres pathologies neurologiques, sont associés à des altérations précoces du développement cérébral, souvent induites in utero et durant la période néonatale. Ces troubles révèlent des dysfonctionnements dans les mécanismes fondamentaux du neurodéveloppement, un domaine clé de nos recherches au sein de Neurochlore. Nos travaux ont étudié le rôle central du GABA et les anomalies de la régulation du chlorure intracellulaire, conduisant à une activité neuronale immature, retrouvée notamment dans les troubles neurodéveloppementaux.
La Bumétanide, un inhibiteur sélectif du transporteur NKCC1, se révèle un candidat thérapeutique prometteur pour restaurer cet équilibre en réduisant les concentrations intracellulaires de chlorure, rendant ainsi au GABA son effet inhibiteur physiologique. Forts de ces principes, nous avons initié des essais cliniques afin d’évaluer l’efficacité et la tolérance de la Bumétanide chez des patients atteints de TSA, dans l’espoir d’apporter une nouvelle approche thérapeutique à ces troubles neurodéveloppementaux.
Évaluation Clinique de la Bumétanide dans les Troubles du Spectre Autistique :
Des essais cliniques menés sur plus de 120 enfants atteints de (TSA) ont exploré l’efficacité de la Bumétanide. Les résultats de deux études de phase II (Lemonnier et al., 2012; 2017) ont montré des effets positifs, renforçant l’intérêt de cette molécule comme traitement potentiel. Plusieurs autres études indépendantes ont confirmé l’efficacité de la Bumétanide dans le traitement des TSA, ouvrant ainsi la voie à des essais cliniques de phase III. Une méta-analyse (Xiao et al) montre que plus de 1000enfants ont bénéficié de ce traitement dans plusieurs pays.
Nos articles sur les phases II.a et II.b :
Neurochlore a conclu un partenariat de licence avec les Laboratoires Servier pour mener un essai clinique de Phase III. Cette phase III, approuvée par les autorités européennes, avaient pour objectif de valider l’efficacité de la Bumétanide dans le traitement des TSA et de préparer la demande de mise sur le marché en Europe. L’étude, divisée en deux sous-groupes d’âge (2-7 ans et 7-18 ans), a inclus 420 enfants TSA recrutés dans 40 centres répartis en Europe, Brésil, Australie et États-Unis. Les participants ont été répartis en deux groupes : un groupe traité à par la Bumétanide et un groupe placebo, sur une période de six mois. La Bumétanide a été administrée sous une forme liquide, approuvée et testée dans les précédentes phases II.
Le détail des études phase III sont accessibles sur les sites :
- Pour l’étude des enfants âgés de 2 ans à moins de 7 ans : https://clinicaltrials.gov/ct2/show/NCT03715153
- Pour l’étude des enfants âgés de 7 ans à moins de 18 ans : https://clinicaltrials.gov/ct2/show/NCT03715166
Toutefois, les résultats de cet essai n’ont pas mis en évidence de différence significative entre le groupe traité et le groupe placebo. Les critères d’évaluation de l’efficacité du traitement n’ont pas atteint le seuil requis, empêchant ainsi la validation de la phase III et la mise sur le marché.
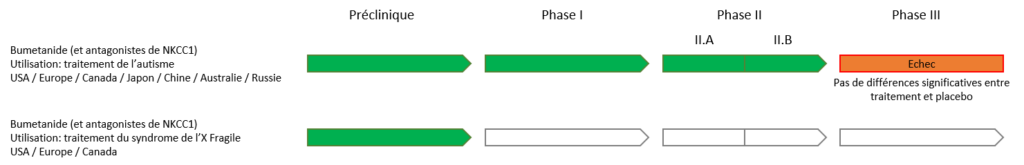
Malgré ces résultats négatifs, le potentiel de la Bumétanide dans le traitement des TSA reste un sujet de recherche active. Les observations des cliniciens ayant participé à l’étude, ainsi que les analyses statistiques, suggèrent que certains sous-groupes de patients pourraient mieux répondre au traitement. Fort de cette hypothèse, Neurochlore a investi dans un nouveau projet visant à identifier ces répondeurs.
